Recent Faculty Publication: Drebrin - a New Player in Angiotensin II-induced Aortopathies
Sawada, H**., B. C. Wright, J. Z. Chen, H. S. Lu*, and A. Daugherty*
August 2018 - no abstract
http://dx.doi.org/10.1093/cvr/cvy205.
Despite major advances in hypertension therapy, even optimally treated hypertensive
patients have high mortality compared to normotensive subjects. One manifestation of
hypertension is aortic remodeling that predisposes to several cardiovascular diseases.
There are still many unknowns on the mechanisms of the cellular and structural
changes of hypertension-induced aortic remodeling. A publication by Zhang and
colleagues in this issue of Cardiovascular Research reports effects of drebrin on
angiotensin II (AngII)-induced aortic remodeling,1 which provides new insights into
understanding mechanisms of vascular diseases.
Drebrin is an actin-binding protein that was identified originally in neuronal cells.2 It has
two isoforms that are splice variants, with drebrin A being expressed in neural cells, and
drebrin E is present in several other cell types, including vascular smooth muscle cells
(SMCs).3 Stiber and colleagues found that drebrin was abundant in atherosclerotic
lesion of humans and mice.6 They also demonstrated increased drebrin expression in
SMCs following wire-injury of the carotid artery in mice. This initial observation provided
a rationale for Zhang et al1 to develop and study mice with SMC-specific deletion of
drebrin.
SMC-specific deletion of drebrin was achieved by breeding drebrin floxed mice with
mice expressing Cre under the control of the endogenous SM22 promoter. The authors
confirmed that there was profound reduction of drebrin in SMCs. Consistent with
previous reports, the SM22 promoter also led to deletion of drebrin in non SMCs
including fibroblasts.4 Deletion of drebrin in SMCs had no effect on AngII-induced
increases in systolic and diastolic blood pressure, as measured by both a tail-cuff based
technique and radiotelemetry. Drebrin deficiency in SMCs also had no effects on AngIIinduced
ex vivo aortic contractility, which occurs only in the infrarenal region of the
aorta.5 Despite the lack of effect of drebrin deficiency on physiological parameters, there
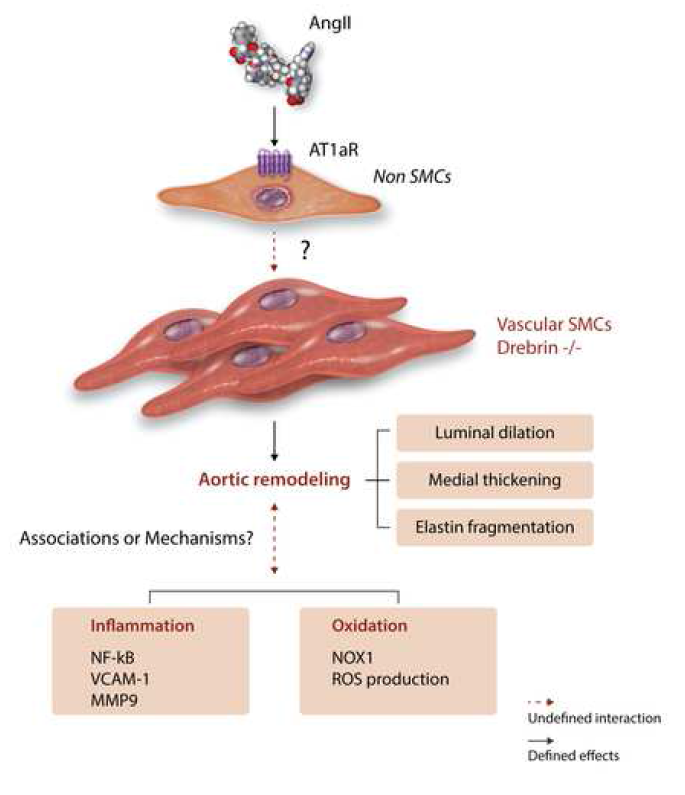
were profound effects on aortic pathology. This included increased AngII-induced aortic
wall thickness, lumen area, and elastin breaks of the excised ascending aorta, and
increased diameter of the aortic sinus in vivo (Figure 1). These changes did not occur in
saline-infused mice. This augmentation of AngII-induced aortic pathology was only
observed in the ascending aorta, not the descending aorta or bronchial arteries.
Previous studies have demonstrated AngII-induced aortic thickening is due to
hyperplasia in the ascending aorta, and hypertrophy in the rest of the aorta.6,7
Therefore, the response in SMC-specific drebrin deleted mice is consistent with a
region-specific effect on hyperplasia.
Having demonstrated the compelling phenotype of drebrin deficiency, subsequent
studies delved into defining mechanisms of this effect. This included measurements of
SMC proliferation, increased collagen I mRNA abundance, phosphorylation of ERK1/2,
enhanced NFκB signaling, increased VCAM-1 expression, increased MMP-9
abundance and enzymatic activity, increased adventitial macrophage accumulation,
increased superoxide, and increased Nox1 mRNA and protein (Figure 1). All these
measurements were performed on tissues extracted from mice infused with AngII for 28 days.
At this interval, there is profound pathology in the ascending aorta. This leads to a
“chicken and egg” situation in terms of mechanistic interpretation: Did the measured
changes promote the pathology? Or were they a consequence of the pathology? Indeed
at this interval of AngII infusion, in addition to processes promoting pathology, they may
be reparative responses in an attempt to “heal” the aorta. There is no clear mode to
resolve this conundrum. One potential approach is to acquire tissues at selected
intervals during the disease evolution to determine whether changes can be observed
prior to the appearance of overt pathology. While potentially enables greater
mechanistic insight, it also greatly ratchets up the difficulty of performing the studies.
One mechanism explored for promoting pathology was the potential for AngII
responsiveness to be enhanced in the affected region. This was examined through
measurement of mRNA abundance of AngII type 1a (AT1a) receptors, which was not
measurably increased by drebrin deletion. Although there has been a pervasive
assumption that AngII-induced aortic pathology is due to direct stimulation of SMCs, this
has not been sustained by experimental approaches. Indeed, several groups have
deleted AT1a receptor from SMCs but failed to detect effects on AngII-induced aortic
pathologies.5,8,9 Consistent with AngII-induced aortic pathology not being attributed to
direct stimulation of AT1a receptor on SMCs, the authors failed to co-precipitate drebrin
and AT1a receptors in HEK-293 cells. These findings implicate that effects of SMCspecific
drebrin on preventing aortic remodeling are not through direct interaction with
AngII activation of AT1a receptors (Figure 1).
This study also begs the question of why drebrin deficiency only influenced AngIIinduced
pathology in the ascending aorta. One potential explanation is the difference of
embryonic origins of SMCs in the ascending and descending aortic regions. The aortic
sinus and ascending aorta are populated with SMCs derived from second heart field
and cardiac neural crest, whereas the descending aorta is populated with SMCs derived
from somites.10-14 Although it is a spatial concordance of these different embryonic
origins with the effect of drebrin deletion on AngII-induced aortic remodeling, there has
no direct evidence that SMCs from these different locations display functional
differences.
Defining sex differences is important, since this has a major effect on many
cardiovascular responses.15 In this study, both male and female mice were studied.
Given the recent clinical data illustrating sex differences in thoracic aortic aneurysm
phenotype in male and female patients, it would be interesting to explore whether
drebrin deletion in aortic remodeling is afforded similarly to male and female mice.
Representing the data in a sex-specific manner may provide further insight into
understanding mechanisms of the regional specific pathology.
In conclusion, drebrin deficiency in SMCs leads to region-specific aortic remodeling in
AngII-infused mice. We look forward to future studies to provide further mechanistic
insight into AngII-induced aortic pathology.
*Saha Cardiovascular Research Center Faculty
** Saha Cardiovascular Research Center Fellow