Recent Faculty Publication: Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo
Moua Yang, Andaleb Kholmukhamedov, Marie L. Schulte, Brian C. Cooley, Na’il O. Scoggins, Jeremy P. Wood*, Scott J. Cameron, Craig N. Morrell, Shawn M. Jobe, and Roy L. Silverstein
Abstract
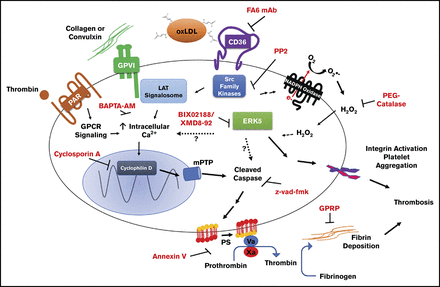
Dyslipidemia is a risk factor for clinically significant thrombotic events. In this condition, scavenger receptor CD36 potentiates platelet reactivity through recognition of circulating oxidized lipids. CD36 promotes thrombosis by activating redox-sensitive signaling molecules, such as the MAPK extracellular signal-regulated kinase 5 (ERK5). However, the events downstream of platelet ERK5 are not clear. In this study, we report that oxidized low-density lipoprotein (oxLDL) promotes exposure of procoagulant phosphatidylserine (PSer) on platelet surfaces. Studies using pharmacologic inhibitors indicate that oxLDL-CD36 interaction-induced PSer exposure requires apoptotic caspases in addition to the downstream CD36-signaling molecules Src kinases, hydrogen peroxide, and ERK5. Caspases promote PSer exposure and, subsequently, recruitment of the prothrombinase complex, resulting in the generation of fibrin from the activation of thrombin. Caspase activity was observed when platelets were stimulated with oxLDL. This was prevented by inhibiting CD36 and ERK5. Furthermore, oxLDL potentiates convulxin/glycoprotein VI-mediated fibrin formation by platelets, which was prevented when CD36, ERK5, and caspases were inhibited. Using 2 in vivo arterial thrombosis models in apoE-null hyperlipidemic mice demonstrated enhanced arterial fibrin accumulation upon vessel injury. Importantly, absence of ERK5 in platelets or mice lacking CD36 displayed decreased fibrin accumulation in high-fat diet-fed conditions comparable to that seen in chow diet-fed animals. These findings suggest that platelet signaling through CD36 and ERK5 induces a procoagulant phenotype in the hyperlipidemic environment by enhancing caspase-mediated PSer exposure.
*Saha Cardiovascular Research Center Faculty