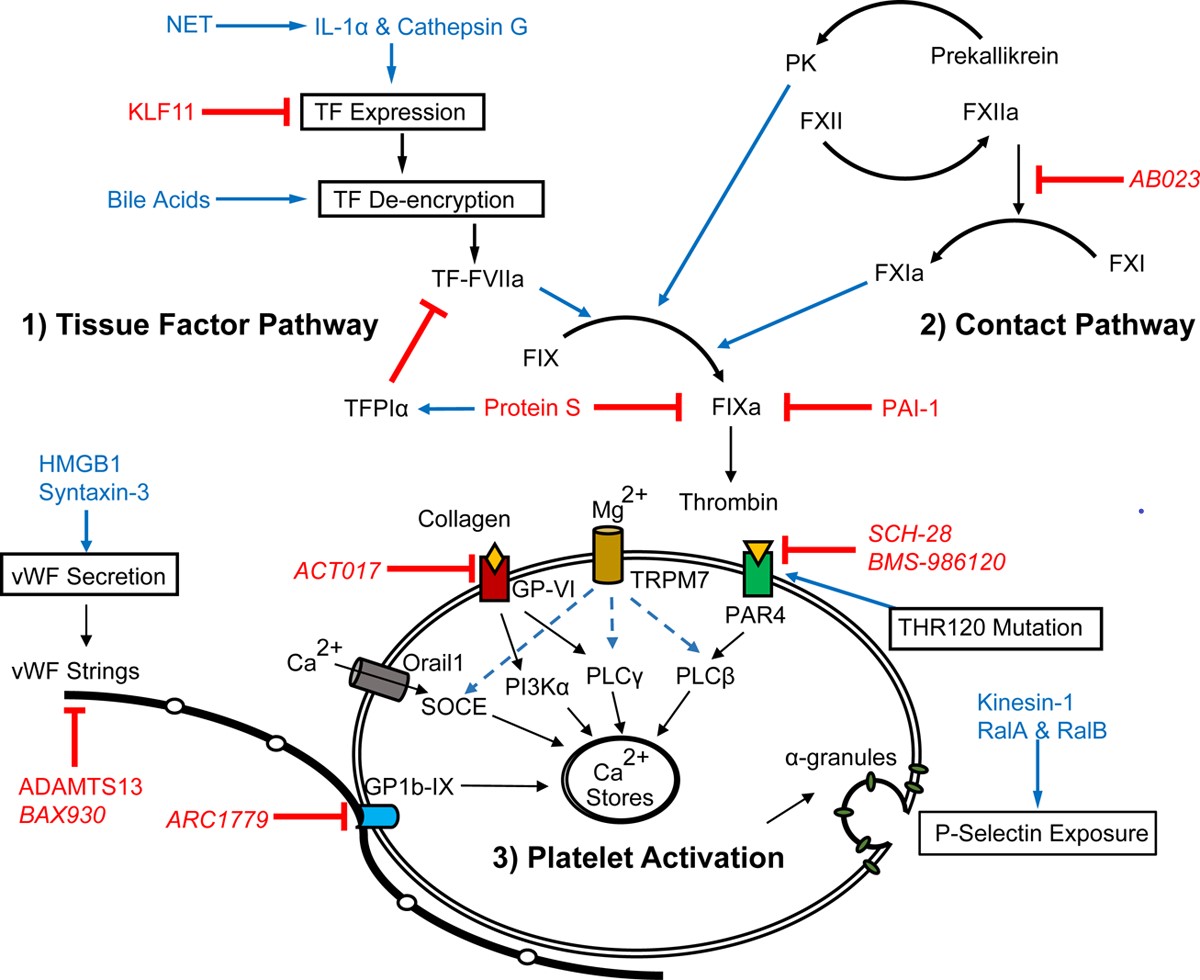
Hemostatic clot formation is the result of a balance between the procoagulant system responding to tissue trauma and the anticoagulant system, which restricts clot formation to the injury site. Imbalances in coagulation lead to a tendency towards either thrombosis or bleeding. Over the past 2 years, studies published in Arteriosclerosis, Thrombosis, and Vascular Biology have provided insights into the regulation of this crucial system. Here, we highlight recent discoveries concerning the 2 pathways of thrombin formation, the extrinsic TF (tissue factor) pathway and the intrinsic contact pathway, and the contributions of platelet activation to the thrombotic process.
Regulation of TF Expression and Activity
TF is a transmembrane glycoprotein cofactor constitutively expressed by subendothelial cells that serve as a high-affinity receptor for and promotes the catalytic activity of FVIIa (factor VIIa).1,2 Upon vascular injury, TF initiates the extrinsic coagulation pathway by binding FVIIa and promoting the activation of FX (factor X) and FIX (factor IX). The importance of TF activity was explored in a recent microfluidic study by Zhu et al,3 which showed that a single molecule of TF can generate up to 92 000 molecules of thrombin and >200 000 fibrin monomers during a 500-second clotting window. They also calculated that the produced thrombin only has a 70 ms half-life. Thus, a single molecule of thrombin is only active long enough to produce 3 fibrin monomers, and robust thrombin generation is required to produce a stable clot.
Many intravascular cells, including neutrophils and monocytes, can be stimulated to express detectable levels of TF.4 These cells might also release TF into the circulation, either as the soluble extracellular form or as full-length protein incorporated into microvesicles.5 Recent studies have identified additional mechanisms regulating TF expression. Neutrophils can release neutrophil extracellular traps, which consist of DNA, histones, and granular enzymes, and Folco et al6 showed that neutrophil extracellular trap-associated effectors induce endothelial cell expression of TF through an interleukin 1α and cathepsin G-dependent pathway. According to a recent study by Liang et al,7 the transcription factor KLF-11 (Krüppel-like factor 11) binds to the F3 promoter to inhibit TF transcription. Under basal condition, endogenous KLF-11 is sufficient to maintain low levels of TF in vascular smooth muscle and endothelial cells. Knockout of TF specifically in vascular smooth muscle cells inhibits thrombus formation in the ferric chloride injury model, while vascular smooth muscle-specific Klf11−/− mice have increased TF and a prothrombotic phenotype. Conversely, overexpression of KLF-11 potently inhibited TNF (tumor necrosis factor)-α-induced TF expression in human aortic smooth muscle cells at both the mRNA and protein levels. Thus, through its regulation of TF, KLF-11 is a key controller of vascular smooth muscle cell procoagulant activity.
Although procoagulant TF is clearly expressed in the vascular subendothelium, it has also been proposed that many different cell types express an inactive, encrypted form of TF that requires de-encryption.8 A recent article by Baker et al9 has provided insight into the regulation of TF de-encryption. Hepatocytes have direct exposure to the plasma because of the fenestrated endothelium of the liver vasculature. Consequently, hepatocytes express an encrypted form of the TF/FVIIa complex. Baker et al9 showed that bile acids stimulate the de-encryption of this TF/FVIIa complex in a murine cholestasis model, possibly explaining the hypercoagulability seen in patients with liver disease. The contribution of microvesicle TF activity has also been explored. Stark et al10 found a clear correlation between cancer-derived microvesicle TF activity and the risk of deep vein thrombosis in prostate cancer, one of the most prothrombotic type of cancers. They also showed that tumor-derived microvesicles can induce thrombosis in animal models. However, microvesicle TF was not sufficient by itself to cause deep vein thrombosis. Clot formation also required synergistic endothelial cell TF and surface exposure of the phospholipid phosphatidylethanolamine but not phosphatidylserine.
Once activated, TF/FVIIa is inhibited by the anticoagulant TFPI-α (TF pathway inhibitor α), which is downregulated in women taking hormonal contraception.11 Recently, Tanratana et al12 showed that reduced TFPI-α in premenopausal women on hormonal contraception results in a 2- to 3-fold increase in the rate of FIX activation and an increase in the amount of circulating FIXa. They also observed an inverse correlation between circulating FIXa and plasma concentrations of TFPI-α and the cofactor PS (protein S). PS promotes TF/FVIIa inhibition by TFPI-α and also directly inhibits FIXa activity.11,13 Plautz et al14 defined the mechanism of this latter function, showing that PS interacts with the heparin-binding exosite of FIXa and that infusion of the FIXa K132A/R170A mutant protein, which cannot bind PS, significantly increases clot formation in hemophilia B mice.
Contributions of the Contact Pathway
FIXa is also generated through the contact pathway of coagulation, a proteolytic cascade initiated by FXII (factor XII), which activates both factor XI (FXI) and the proinflammatory kallikrein-kinin system.15 Upon exposure to negatively charged surfaces (eg, DNA) or pathogens (eg, long-chain polyphosphates), FXII undergoes a conformational change and produces FXIIa (activated FXII). Subsequently, FXIIa activates the intrinsic coagulation system by FXI (factor XI) cleavage to form FXIa (activated FXI). FXIa then proteolytically activates FIX. FXIIa also digests prekallikrein to release PK (plasma kallikrein), which reversely activates FXII to produce an enhanced positive feedback activation loop.15
Multiple lines of evidence have shown that FXII is dispensable for hemostasis but promotes thrombotic processes, such as venous thromboembolism, suggesting this pathway as a viable therapeutic target.15 Lorentz et al16 showed that AB023, an antibody which selectively blocks FXI activation by FXIIa and demonstrates a dose-dependent duration of limited anticoagulation in mice, has antithrombotic activity in a baboon model but does not increase bleeding time. They then performed a first-in-human study of the antibody. They reported a prolonged activated partial thromboplastin time in patients treated with the antibody, demonstrating its efficacy, and confirmed that the antibody does not impair either thrombin-mediated FXII activation or FXIa enzymatic activity. Most importantly, they reported no severe adverse events were observed, with only one patient experiencing a bruise at the injection site.
Similar to the TF pathway, the contact pathway is tightly downregulated in vivo, by C1 esterase inhibitor, which targets both FXIIa and PK.15 Puy et al17 showed that the endothelial serine protease inhibitor PAI-1 (plasminogen activator inhibitor-1) also downregulates the contact pathway. Specifically, PAI-1 forms an inhibitory complex with FXIa, which is then either shed into circulation or endocytosed by endothelial cells, trafficked through endosomes and lysosomes, and degraded. Promoting these natural suppressive mechanisms represents a potential therapeutic strategy to prevent thrombosis through contact pathway inhibition.
In addition, the contact pathway influences the host response to a variety of pathological processes besides thrombosis, including atherosclerosis, stroke, and sepsis.15 Zilberman-Rudenko et al18 demonstrated that long-chain polyphosphates released by bacterial pathogens induce platelet activation and fibrin generation in vivo through a FXII-dependent mechanism and that pretreatment with FXIa antibody suppresses platelet and fibrin consumption in a bacterial sepsis model.19 Meanwhile, data also suggest cross-talk between the proinflammatory and procoagulant processes.20 Visser et al21 found that PK, the major proinflammatory downstream mediator of FXIIa, also contributes to FIXa activation by FXIIa, a process which operates parallel to the FXIa pathway. The ability of PK to activate FIXa could be explained by the high sequence homology between the active sites of PK and FXIa, a well-described FIX activator.22
Platelet Activation
Whether generated through the TF or contact pathway, the final substrate thrombin amplifies its own production, in part, through the activation of PAR-1 and PAR-4 (protease-activated receptors 1 and 4) on platelets and generation of a platelet surface capable of supporting prothrombinase assembly.23 PAR-1 activity is inhibited by the antiplatelet agent vorapaxar,24 and a recent study by Tourdot et al25 has highlighted the importance of PAR-4 activation in thrombosis. This group previously identified a variant in PAR-4 (PAR-4-Thr120), which is prevalent in blacks and accounts for ≈50% of the platelet hyperactivity observed in this group.26 They have now shown that platelets expressing PAR-4-Thr120 have increased Gq and G13 activation in response to thrombin, undergo a more dramatic shape change, and produce larger clots under flow. These data suggest that PAR receptors, such as PAR-4, may be valuable targets for antithrombotic therapy, particularly in individuals with the Thr120 variant.
Two recent publications have assessed the therapeutic potential of anti-PAR-4 agents. First, Wilson et al27 studied the effects of BMS-986120, a reversible PAR-4 antagonist, on ex vivo platelet thrombus formation in a Phase I clinical trial. They found that platelets from BMS-986120-treated patients had dramatically reduced responses to PAR-4 stimulation and formed smaller clots under high shear flow conditions, which were similar in size to those formed by platelets from patients treated with aspirin or a combination of aspirin and clopidogrel. Second, Lin et al28 showed that the nonanticoagulant heparin SCH-28 specifically blocks thrombin-mediated PAR-4 activation and reduces platelet thrombus formation under flow and suggest that SCH-28 may be a safer alternative to traditional heparin therapy, as it does not promote FXa or thrombin inhibition by antithrombin.
Platelets are also activated through the collagen receptor GP-VI (glycoprotein VI).29 This interaction is often described as the initial activator of platelets, as collagen is exposed at the subendothelial site of injury. Recent work by Lehmann et al30 has described an important role for GP-VI in clot progression, in a model for venous thromboembolism. They developed a microfluidic system to mimic the low-flow conditions of a venous valve. In this system, they found that TF activity promotes fibrin deposition, platelets adhere to the fibrin and expose phosphatidylserine, and additional platelets are recruited to promote thrombus growth. This final step was dependent on GP-VI, as it could be inhibited by the anti-GP-VI antibody Fab fragment ACT017 or by the fibrin fragment d-dimer, which binds GP-VI. GP-VI is also being actively pursued as a therapeutic target. Voors-Pette et als31 have reported the results of a first-in-human study of ACT017. This was a safety study in healthy volunteers, and the authors reported no serious adverse events, no change in platelet count, no change in GP-VI expression on platelets, and no change in the concentration of shed, soluble GP-VI in plasma.
New studies have also elucidated the signaling pathways that lead to platelet activation in response to either thrombin or collagen. Adam et al32 knocked out the motor protein kinesin-1 heavy chain in mice and observed a re-bleeding phenotype following tail clip and thrombus instability in the ferric chloride injury model. They subsequently showed that the platelets responded less to low-dose thrombin or collagen stimulation, defined by reduced aggregation, ATP and PF-4 (platelet factor 4) secretion, and P-selectin exposure. These deficiencies could be overcome by stimulation with high concentrations of thrombin. Thus, kinesin-1 is a component of the thrombin signaling pathway in platelets. In addition, work by Wersäll et al33 identified a specific role for the Ral family of small GTPases. They knocked out either RalA or RalB or both in a mouse model and observed reduced P-selectin exposure in response to either thrombin or collagen, although total P-selectin was actually slightly increased. In contrast, they saw no change in activation of integrin αIIbβ3, secretion of α-granule cargos PF-4 and TGF-β, or ex vivo thrombus formation. Although both RalA and RalB contributed to P-selectin exposure, the most dramatic phenotype was observed in double-knockout mice. However, they did observe decreased formation of platelet-leukocyte aggregates, a P-selectin-dependent process. This work shows that P-selectin exposure is regulated differently than α-granule release and is not essential for thrombus formation. Finally, Laurent et al34 used knockout mice and inhibitors to show that PI3K-α (phosphoinositide 3-kinase α) is involved in platelet activation in response to low doses of collagen. Unlike kinesin-1 and Ral GTPases, PI3Kα was not involved in thrombin signaling. In response to collagen, knockout or inhibited platelets had reduced aggregation, dense granule secretion, and adhesion to immobilized VWF (von Willebrand factor). The mice also had reduced thrombus formation in a laser injury model but did not show any impairment in the tail clip bleeding assay.
Platelet activation through GP-VI, PARs, and other receptors requires release of intracellular calcium stores into the cytosol.35 Recent work by Gotru et al36 showed that these stores are regulated by TRPM-7 (transient receptor potential cation channel, subfamily M, member 7), a protein which they had previously identified as a regulator of magnesium in megakaryocytes.36,37 Platelets from TRPM7−/− mice exhibit decreased calcium release, aggregation, P-selectin exposure, and integrin activation in response to either collagen or thrombin. In addition, the authors observed reduced thrombus formation ex vivo in a whole blood flow system and showed that the mice were protected from ferric chloride-induced thrombosis and from cerebral ischemia, while there was no change in the tail bleed assay. Lopez et al38 added to these observations by showing that calcium signaling is regulated by the calcium sensor STIM-1 (stromal interaction molecule 1) and by the phosphorylation of Ser2152 on the cytoskeletal protein filamin A, which promotes its interaction with STIM-1. They showed that inhibition of the STIM-1-filamin A interaction enhances calcium release and platelet aggregation. Collectively, these studies have identified several new potential downstream therapeutic targets, which could selectively inhibit specific aspects of platelet function to prevent thrombosis.
Other Regulators of Platelet Function
Platelet function is also influenced by several other factors, including the adhesive protein VWF and flow forces. Platelets bind to VWF through the glycoprotein IB-IX-V receptor complex.39 Chen et al40 showed that by blocking VWF binding with the aptamer ARC1779, they can improve the ex vivo stability of stored platelets. They found that refrigerated murine and human platelets incubated with ARC1779 had increased recovery and half-life post-transfusion. They also had increased hemostatic function, evidenced by a reduced clot time in the tail bleed model, suggesting that VWF binding is at least partially responsible for the loss of activity and lifetime that has been observed with cold-stored platelets. Abdelgawwad et al41 used a different approach to show the potential therapeutic benefit of blocking the platelet/VWF interaction. Thrombotic thrombocytopenic purpura is caused when congenital or acquired loss of the enzyme ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats 13) results in an increase in circulating high molecular weight VWF multimers.42 Abdelgawwad used the natural endocytic machinery to load platelets with ADAMTS13 and showed that these platelets have antithrombotic properties when transfused into ADAMTS13−/− mice or added into whole blood from thrombotic thrombocytopenic purpura patients, supporting this process as a novel treatment approach.41 Adili and Holinstat43 similarly assessed the efficacy of introducing ADAMTS13 to treat thrombotic thrombocytopenic purpura. They showed that ADAMTS13−/− mice are prone to VWF-dependent microvascular thrombi in the brain, which are independent of fibrin, and that infusion of BAX930, a recombinant form of ADAMTS13, was able to resolve these thrombi.
VWF is stored in Weibel-Palade bodies of endothelial cells and secreted into plasma.44 Recent work from Biswas et al45 showed that VWF secretion is promoted by high-mobility group box 1, and this process is dramatically enhanced by the presence of polyphosphate chains in the same size range as those secreted by activated platelets. The released VWF then supports platelet adhesion and formation of platelet/VWF strings on the endothelial surface, suggesting a positive feedback loop between platelets and VWF in this process. Intracellularly, Schillemans et al46 identified syntaxin-3 as a key mediator of the membrane fusion process that allows for VWF secretion. Syntaxin-3 functions with vesicle-associated membrane protein 8, a well-described exocytosis regulator, to control this process, but has no impact on Weibel-Palade body formation. VWF is also found in platelet α-granules, but interestingly, Doddapattar et al47 found that only endothelial VWF promotes platelet adhesion and subsequent inflammation and leukocyte adhesion in the ApoE−/− atherosclerosis model, suggesting that VWF may serve different functions depending on its source.
It has long been understood that shear forces regulate VWF activity and platelet aggregation, and this appears to be particularly true for turbulent flow.48 According to the work of Bortot et al,49 VWF activity is specifically regulated by turbulent flow patterns. Bortot compared laminar, transitional, and turbulent flow patterns and observed that turbulent flow resulted in the greatest loss of VWF multimers (indicating the highest activity of ADAMTS13) and the lowest VWF activity, as measured by its ability to bind platelets and collagen.49 These data offer a possible mitigating factor to prevent spontaneous thrombosis in venous valves, where turbulent flow patterns are prevalent.34 Anyanwu et al50 described another potential mitigating factor, the ectonucleotidase CD39. They showed that CD39−/− mice have increased thrombus size in a stasis injury model, increased P-selectin exposure and VWF levels, and increased platelet-leukocyte aggregates. Thus, the ability of CD39 to hydrolyze ATP and ADP appears to protect from venous thrombosis.
The pathological environment also has the power to shape platelet phenotype and function and can impact the efficacy of antiplatelet therapies, such as aspirin and clopidogrel.51 Cameron et al52 demonstrated that platelets isolated from patients with vascular and metabolic diseases, including patent ductus arteriosus, diabetes mellitus, and hypertension, have higher activity and are resistant to antiplatelet agents, when compared to platelets isolated from healthy volunteers. The mechanism underlying this change in platelet phenotype is not clearly understood but is partly mediated by extracellular regulated protein kinase 5.51,52
Summary
Over the past 2 years, studies published in Arteriosclerosis, Thrombosis, and Vascular Biology have improved our understanding of the regulation of thrombin generation, through both the TF and contact pathway, and of platelet activation and function (Figure). These studies have identified new pathways and targets, which may lead to improved anticoagulant and antithrombotic agents in the years to come.
Figure. Recent advances have been made in our understanding of (1) TF (tissue factor) expression, de-encryption, and downstream substrate FIXa (activated factor IX) activation; (2) regulation of the contact pathway and its crosstalk with inflammation; and (3) receptors, signaling, and extracellular components that regulate platelet activation. Blue text indicates promoters, red indicates inhibitors, and italics indicate therapeutic agents. GP-VI indicates glycoprotein VI; IL, interleukin; KLF-11, Krüppel-like factor 11; NET, neutrophil extracellular trap; PAR, protease-activated receptor; PK, plasma kallikrein; TFPI-α, tissue factor pathway inhibitor α; and VWF, von Willebrand factor.